The development of new materials
and the improvement of the properties of conventional materials
have been based on the results of many experiments conducted
by changing the chemical composition and the manufacturing conditions;
empirical rules and phenomenology form the basis for this development
and improvement. Although these have mainly been adopted for
material development, a quite different technological approach
has recently been developed. This new technology involves forming
the structure of a metallic material with desirable properties
based on theoretical assumptions from the structure of atoms
and crystals. The manufacturing conditions for a material with
that structure are then theoretically investigated. This procedure
is conducted by computer simulation, and is called computational
metal physics or computational materials science.
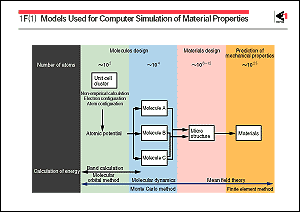
The figure shows the models that are mainly used at present,
classified in accordance with the quantity of atoms of the relevant
system. From the most microscopic standpoint, the energy level
of the electrons of one atom or atoms within a unit lattice is
first analyzed by quantum mechanics. Next, when dealing with
the interaction or migration of a large number of atoms, it is
analyzed by molecular dynamics, the Monte Carlo method, or some
other method. These analyses enable such properties as the lattice
parameters, elastic modulus, specific heat, and electrical conductivity
to be presumed, as well as structural information such as lattice
defects, the grain boundary and surface, and transport properties
such as diffusivity and viscosity. In the macroscopic interpretation,
the mean field theory based on thermodynamics and a finite-element
method are applied. The present level of computational materials
science is already sufficient to explain some of the phenomena
that are empirically known, and great progress with this science
can be expected in future. |
|
 |
 |
 |
|
|